- Introduction to medical devices standards
- What is harmonization and what is the status of harmonization with MDR?
- So, what has changed?
- Lessons learned from recent submissions to NBs
- Common mistakes made during medical device development
Introduction to medical devices standards
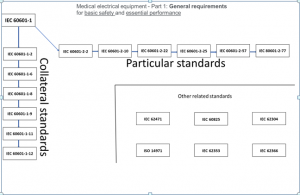
BASIC SAFETY
Freedom from unacceptable RISK directly caused by physical HAZARDS when ME EQUIPMENT is used under NORMAL CONDITION and SINGLE FAULT CONDITION
ESSENTIAL PERFORMANCE
Performance of a clinical function, other than that related to BASIC SAFETY, where loss or degradation beyond the limits specified by the MANUFACTURER results in an unacceptable RISK
MEDICAL ELECTRICAL EQUIPMENT (ME EQUIPMENT – medical device)
Electrical equipment having an APPLIED PART or transferring energy to or from the PATIENT or detecting such energy transfer to or from the PATIENT and which is:
- Provided with not more than one connection to a particular SUPPLY MAINS; and
- Intended by its MANUFACTURER to be used:
(a) In the diagnosis, treatment, or monitoring of a PATIENT; or
(b) For compensation or alleviation of disease, injury or disability - Know your standards
- Start with terms and definition
- Until you know terms and definitions by heart always keep them open while reading
- Ask testing houses for guidance
- Not everything intended for hospital use should be certified towards IEC 60601-1
- Talk to NBs
- Perform FDA pre-submissions
What is harmonization?
- IEC and ISO standards turned EN
- Are very similar to international standards
- Explicitly published in the EU Official Journal
- These standards are uniform throughout Europe
In short, all harmonized standards which are used should cover the Essential Requirements. If gaps still remain, they must be filled with other verification.
![]()
![]()
What is the status of harmonization with MDR?
In June 2019, the European Commission issued the draft standardization request as regards medical devices in support of Regulation (EU) 2017/745 and in vitro diagnostic medical devices in support of Regulation (EU) 2017/746.
This request identified the first batch of standards for consideration for harmonization to the MDR and the European In Vitro Diagnostics Regulation (IVDR). There are a large number of standards in the mandate:
- Some 57 revisions and six totally new standards for the MDR.
- 30 revisions and four new standards for the IVDR.
- With some overlap, there’s a total of 79 separate documents in the work program.
Priorities are being given to a handful of key standards, specifically those on quality systems (ISO 13485), risk management (ISO 14971), biocompatibility (ISO 10993-1), clinical trials (ISO 14155), and labelling (ISO 15523), including labelling for devices containing phthalates (ISO 15986).
So, what has changed?
On the paper – nothing.
In practice the NBs are scared of the transition and preferring to be on the safe side.
Some are even trying to be more righteous than the Pope.
Sorts of outrages requests such as:
- Medical Device with peristaltic pump should comply with Machinery Directive
- Device should comply with international standard currently in final draft stage.
- Rule 11 – All active devices intended to administer and/or remove medicines, body liquids or other substances to or from the body are in Class IIa, unless this is done in a manner:
— that is potentially hazardous, taking account of the nature of the substances involved, of the part of the body concerned and of the mode of application in which case they are in Class IIb.
Lessons learned from recent submissions to NBs.
- Be very careful when consulting the current directives’ harmonized standards list on the Europa website. For every standard you expect to use, check carefully if there is a later version published by ISO or IEC, and consider using that instead.
- Don’t assume that anything on the current Medical Device Directive (MDD), In Vitro Device Directive (IVDD), and Active Implantable Medical Device (AIMD) lists of harmonized standards will carry over to the MDR/IVDR. That should be verified.
- If there is a discrepancy, talk to your Notified Body about it. Check that they are comfortable with you going to the later version (or, if you have test data against the harmonized version, check that they will accept it).
- Keep an eye on the ISO website, so you can see what changes are coming.
- Finally, you should also have communication with your national standards body about what is happening. There will be local experts who are involved in the development of new ISO documents. They should be able to tell you what’s planned and when new standards are likely to be published.
Lessons learned from recent submissions to NBs.
Use latest EN version of 14897 there are some very important deviations from the international version.
(requirements from the international version are in bold)
- Users shall be informed about the residual risks. This indicates that the information given to the users does not reduce the (residual, some say not only residual) risk any further.
residual risk is defined as the risk remaining after application of risk control measures.
regards “information for safety” to be a control option.
Should you use BS EN ISO 14971:2019 or the older version?
According to BSI shop BS EN ISO 14971:2019 replaces BS EN ISO 14971:2012, on the other side it is not harmonized.
As I see it – it is part of the state of the art and the last revision should be used. Some very important definitions have been added. One of them is term Benefit.
- More attention is given to the expected benefits of using the medical device. The term benefit-risk analysis has been aligned with terminology used in some regulations
- The method for evaluating the overall residual risk and the acceptance criteria are to be defined in the risk management plan
- The review before commercial distribution of the medical device concerns the execution of the risk management plan. The results of the review are documented as the risk management report.